Is your organization structurally capable of a defensible AI decision?
 By
By
Three years of pharma AI conversations have been about what the tools can do. The regulatory record is starting to ask a different question, and most organizations are not ready to answer it.
Pharma has spent three years evaluating AI vendors. Almost none of that evaluation has touched the organization doing the buying.
Walk through any AI conversation in this industry right now, and the same sequence plays out, whether the room is talking about discovery, clinical trials, regulatory review, validation, manufacturing, or pharmacovigilance. A vendor demonstrates a capability. A team scores it against a requirements list. A contract gets signed. The capability is genuine. The demonstration is accurate. What seldom gets asked is whether the organization on the other side of that contract can defend the decision the tool produces.
Where the product-led pattern shows up
Discovery. Platforms like Insilico Medicine and Atomwise generate molecule candidates faster than any prior method. The pitch is speed. The unasked question is who signs off on a candidate an algorithm proposed, and what they would say to a regulator asking why.
Clinical trials. Synthetic control arms, built from real-world data through efforts like Project DataSphere, reduce the need for placebo groups. The pitch is fewer patients exposed to unnecessary risk. The unasked question is whether the statistical reasoning behind that substitution is documented well enough to survive an inspection five years later, after the person who built the model has moved on.
Regulatory review. NLP tools that help EMA and FDA reviewers parse submission data faster are a genuine efficiency gain. They are also training an industry to expect machine-assisted judgment inside the review process itself, ahead of any public standard for how that judgment gets documented.
Validation. Tools like ValGenesis and Kneat are adding AI modules to streamline documentation and risk assessment under the FDA's Computer Software Assurance approach. CSA governs the assurance methodology. It does not answer who owns the risk determination an AI module recommends, or whether that determination would survive a regulator's challenge.
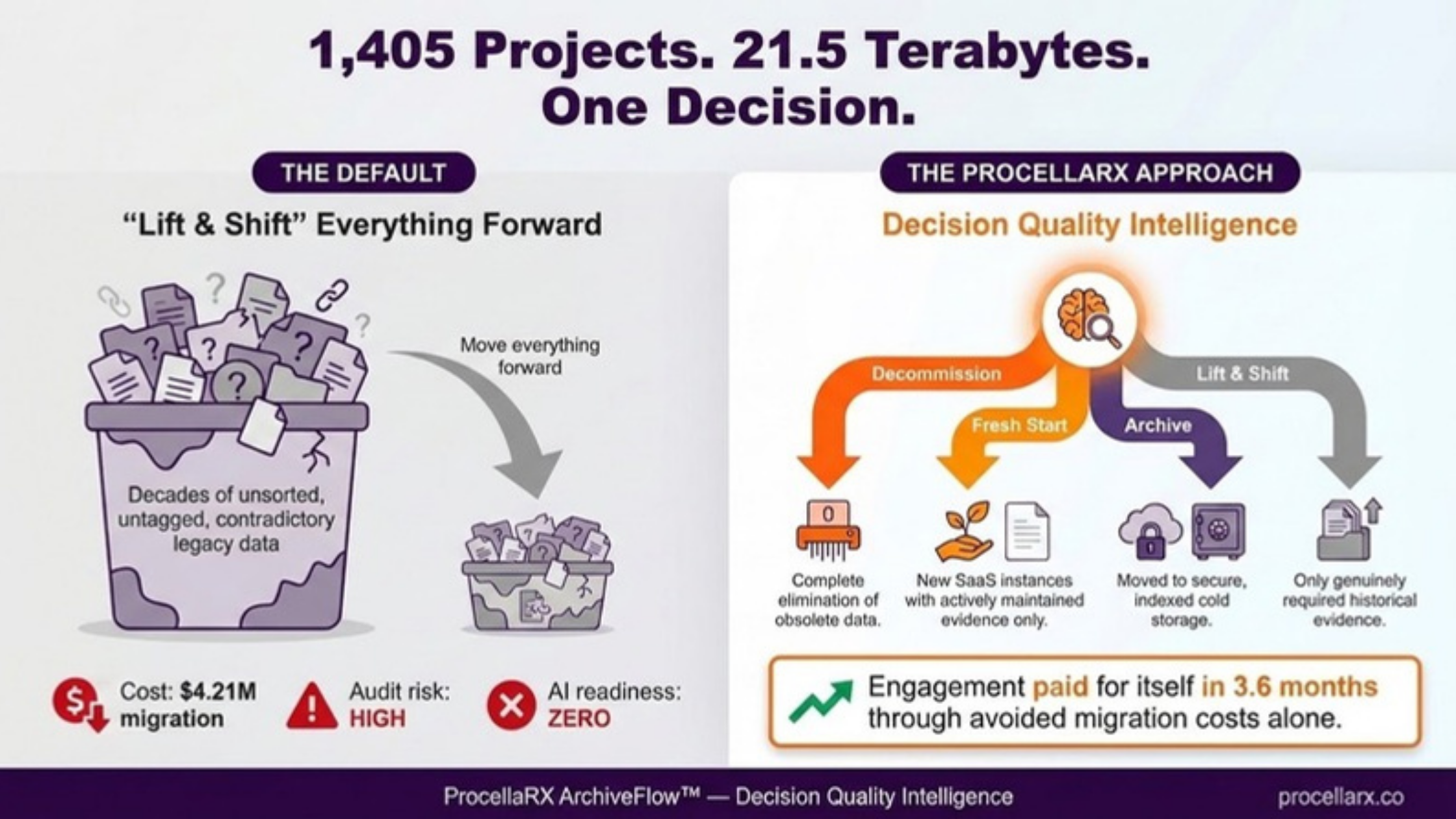
Manufacturing. Predictive maintenance and AI-driven CAPA analysis are cutting downtime and catching deviations earlier. Adoption is already high: internal estimates put predictive maintenance at 85 percent of manufacturing and quality processes, audit readiness support at 82 percent, deviation detection at 78 percent, automated batch record review at 72 percent, and CAPA analysis at 68 percent. Adoption at that scale means fewer humans are reviewing the reasoning behind each flag, at the exact moment more consequential calls are being made faster.
Pharmacovigilance. Systems like Oracle Argus Safety use machine learning to triage adverse event cases and classify seriousness. Speed here can save lives. It can also mean a seriousness classification ships before anyone traces why the model called it that way.
Six domains. Six vendors answering the same question well. None of them are positioned to answer the one underneath it.
The question underneath the product question
For three years, the pharma AI conversation has been product-led. Better molecule generators. Better trial design tools. Better assurance modules. The organizations that get this right over the next three years will not be the ones that bought better tools. They will be the ones that built better governance conditions underneath them.
Is your organization structurally capable of a defensible AI decision? Not whether the AI works. Not whether the vendor is reputable. Whether your organization, today, could answer who made the call, what they knew when they made it, and why it was reasonable given both, and could answer that the same way regardless of who happens to be in the room.
What AI governance in pharma requires
ProcellaRX names this discipline Decision Quality Intelligence, and it rests on three conditions. Every consequential AI-assisted decision either meets all three or it is not, by this standard, defensible.
Conscious. The decision is named and actively made, not inherited from a default setting or a vendor's out-of-the-box configuration. Someone can say, specifically, that this decision was made, and when.
Defensible. The reasoning is captured alongside the outcome, not just the outcome itself. An auditor, a regulator, or a successor two years from now can reconstruct why the call was reasonable, using evidence that existed at the time it was made.
Continuous. The decision is revisited on a defined schedule or trigger, not treated as settled the day the system went live. Quality is designed into the lifecycle, not inspected in after the fact.
Most pharma organizations can point to documentation. Documentation is not the same as reasoning. A validation binder can be complete and still fail this standard if it records what was decided without recording why.
Why the regulatory record already agrees
This is not a ProcellaRX opinion sitting ahead of where regulators are. Three separate developments over the past year point at the same target: the decision behind the output, not the output's polish.
FDA finalized its Computer Software Assurance guidance in September 2025 and issued an update in February 2026. The old approach scripted every test in advance regardless of what was at stake. The new one asks manufacturers to size their testing to the risk of the specific function and to write down the reasoning for that sizing decision, not only the result. A manufacturer can no longer show up with a passing test report and call it done. The report has to come with an explanation an inspector can follow.
FDA and EMA followed that in January 2026 with ten jointly authored Guiding Principles of Good AI Practice in Drug Development, the first time the two agencies have put out a shared position on AI across the full product lifecycle. Nothing in the ten principles is enforceable yet. What they establish is a direction: oversight and accountability attached to every stage where an AI system touches evidence a regulator might later rely on.
EMA's Annex 22, covering AI inside GMP manufacturing, has not reached that stage yet. It remains a draft. A multistakeholder workshop wrapped on July 1, 2026, and EMA has indicated a final version will follow later this year. The draft's substance has already moved once in response to industry pushback, so treat any specific provision as unsettled. What has not moved is the underlying ask: documented intended use, a bias check before deployment, and a named threshold for anything touching a critical decision. Three regulators, three different documents, one consistent question. Who is accountable, and can they prove it?
The gap that doesn't show up in a vendor demo
Most organizations can reconstruct, in detail, why a decision failed. Post-incident reviews are a mature discipline in this industry. Ask the same organization to reconstruct why a decision that worked was reasonable at the time, using only evidence that existed then, and the exercise gets much harder.
That asymmetry is the tell. An organization that can only defend its failures has not built decision governance. It has built incident response and called it enough.
Three questions worth asking before the next AI contract
Who owns this class of decision, and do they know it? Not the department. A person, or a defined role, who could be asked directly and would recognize the question as theirs to answer.
Could someone reconstruct the reasoning in twelve months? Using evidence that existed at the time the decision was made, not evidence assembled afterward to justify it.
Does the same answer come back twice? If the outcome depends on which specific person happens to be reviewing the case that week, the organization has a skilled individual. It does not yet have a governed capability.
An organization that answers all three, consistently, is ready to have a serious conversation with any AI vendor. An organization that cannot has a governance gap, and no product roadmap closes it on its own.
Buy the product. Build the capability first.
Vendor conversations in pharma AI will continue at the current pace, and most of them will keep being useful. The demos are not the problem. The absence of a parallel conversation, about whether the buying organization can defend what the tool produces, is the problem.
The Reinvention Lab convenes pharmaceutical and vendor leaders specifically to work through this kind of organizational readiness gap together, in the open, using case material instead of theory. If the three questions above raised more uncertainty than confidence, that is the conversation worth having next.